
21世纪是生物的世纪,生物数据的增长速度越来越快。很多分析工具在开发时并没有考虑到大规模数据的应用场景。在数据量不大的时候,这些工具的计算时间并不会太长,可以让人接受。但在数据规模庞大时,可能就 hold 不住,等待时间让人发指。
加速大规模生物数据的分析速度有很多方法,其中利用 GPU 加速基因组研究是一个非常火的方向。典型的一个例子是 NVIDIA Clara Parabricks,它的 GPU-based GATK4 Best Practice Pipeline 可以比 CPU 版本快 35 到 50 倍。
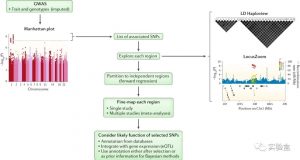
今天要介绍 GWAS_Flow这个工具,它是基于 TensorFlow 的 GWAS 框架,使用的是 EMMAX 方法。
安装
GWAS_Flow 可以基于Git和 Anaconda 安装。它虽然也有 Docker 和 Singularity 版,但目前还在开发,可能会有些问题,不建议使用。
这里使用 Anaconda 安装:
# 克隆 git 仓库
git clone https://github.com/Joyvalley/GWAS_Flow
# 创建conda环境,安装需要的包:
conda create -n gwas_flow python=3.7.3
conda activate gwas_flow
conda install -y tensorflow==1.14
conda install -y scipy pandas numpy h5py
conda install -y -c conda-forge pandas-plink
conda install -y -c conda-forge matplotlib
pip install limix
输入数据
GWAS_Flow 至少需要一个基因型文件和表型文件,如果有 kinship matrix、协变量等,也可加进去。如果没有提供 kinship matrix,GWAS_Flow 会自动使用 van Raden 的方法计算 kinship。
基因型文件
可以接受 HDF5 、CSV 和 Plink(bed/bim/fam) 三种格式。其中,CSV格式是基因型编码为 0,1,2 的 csv 文件,第一行为 header,第一列为样本id,后续每一行是一个样本、每一列是一个 SNP。
表型文件
CSV 格式,第一列是样本 id,第一行是 header。
![图片[1]-GWAS_Flow:使用GPU加速大规模数据的全基因组关联分析 - 生物信息-实验盒](https://www.shiyanhe.com/wp-content/uploads/2020/06/GWAS_Flow1.png)
Kinship文件
可以是 h5py 格式,也可以是 CSV 格式。CSV 格式的Kinship matrix,第一行要加入 header,第一列加入样本 id。
运行
使用 plink 格式的示例数据(输入 plink 格式的文件时,注意要使用文件前缀名再加 .plink 的形式):
python gwas.py -x gwas_sample_data/my_plink.plink -y gwas_sample_data/pheno2.csv -k gwas_sample_data/kinship_ibs_binary_mac5.h5py
命令中的标签和选项包括:
-x , --genotype : file containing marker information in csv or hdf5 format of size
-y , --phenotype : file container phenotype information in csv format
-k , --kinship : file containing kinship matrix of size k X k in csv or hdf5 format
-m : name of column to be used in phenotype file. Default m='phenotype_value'
--cof: file with cofactor information (only one co-factor as of now)
-a , --mac_min : integer specifying the minimum minor allele count necessary for a marker to be included. Default a = 1
-bs, --batch-size : integer specifying the number of markers processed at once. Default -bs 500000
-p , --perm : perform n permutations
--plot : create manhattanplot
-o , --out : name of output file. Default -o results.csv
-h , --help : prints help and command line options
如果要加入协变量,可以使用 --cof 标签:
python gwas.py -x gwas_sample_data/G_sample.csv -y gwas_sample_data/Y_sample.csv -k gwas_sample_data/K_sample.csv --cof gwas_sample_data/cof.csv
如果要进行 permutation,可以加入 --perm n 标签,其中 n 是 permutation 的次数。
如果要输出曼哈顿图,可以加入 --plot True 标签。
总结
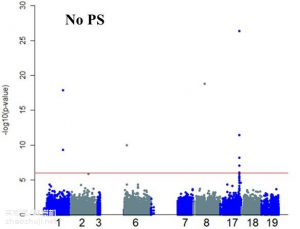
如果数据量小,使用 GPU 速度反而可能会更慢。但当数据量大起来,GPU 带来的提升就很大。比如下面这个图,是 10000 SNPs 分别用 i9 CPU 16核、Tesla P100 的模拟运算,当表型数量超过 800 后, GPU 版的运算时间增加不大,提升效果明显。
![图片[2]-GWAS_Flow:使用GPU加速大规模数据的全基因组关联分析 - 生物信息-实验盒](https://www.shiyanhe.com/wp-content/uploads/2020/06/GWAS_Flow2.png)
GWAS_Flow 目前还不是很完善,但总的来说提供了一个不错的参考框架,可以参考它来开发自己感兴趣的东西。
![图片[3]-GWAS_Flow:使用GPU加速大规模数据的全基因组关联分析 - 生物信息-实验盒](https://www.shiyanhe.com/wp-content/uploads/2020/05/qrcode_shiyanhe.jpg)













暂无评论内容