介绍
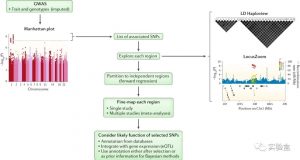
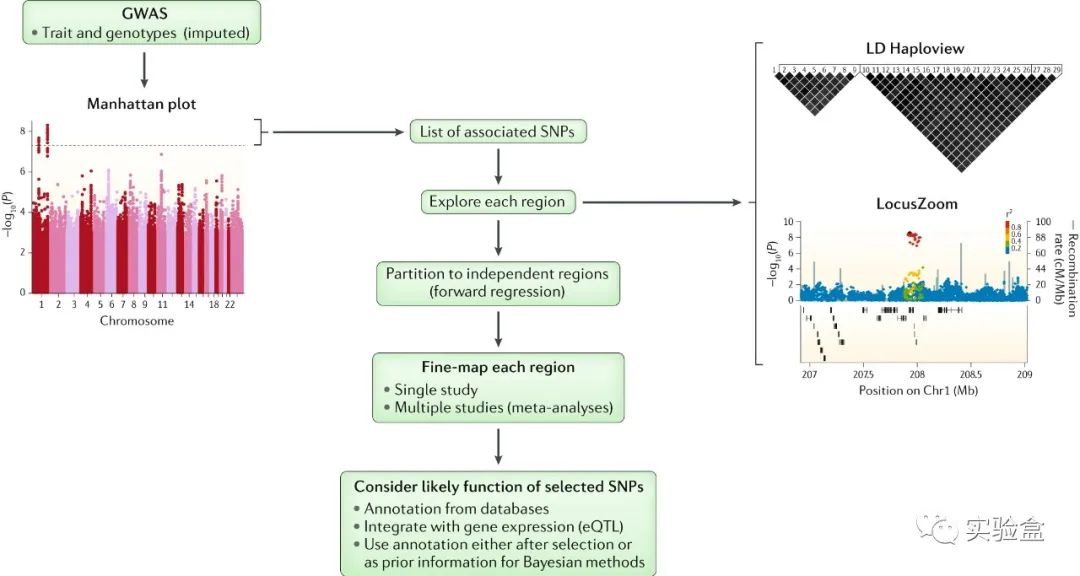
全基因组关联分析(GWAS)是非常流行的定位表型或疾病遗传位点方法。不过很多情况下,GWAS 发现的最显著的 SNP(top SNP 或者 index SNP)并不是真正造成影响的causal SNP(因果SNP),而是因为跟 causal SNP 之间存在的 LD 而变得显著。因而,后续还需要对结果进行 fine-mapping(精细定位),把 causal SNP找到。
如果想了解更多 fine-mapping 的知识,推荐看看 Nature Reviews Genetics From genome-wide associations to candidate causal variants by statistical fine-mapping 这篇综述。
![图片[1]-FINEMAP:使用GWAS摘要数据进行无功能注释数据的精细定位(Fine-mapping) - 生物信息-实验盒](https://www.shiyanhe.com/wp-content/uploads/2020/07/finemap1-1024x546.jpg)
方法
常用的 fine-mapping 方法有 PAINTOR、eCAVIAR等。这些方法利用了 Roadmap、ENCODE 或者 GTEx 的功能注释数据,效果不错。然而,这些方法一般需要其他组学注释数据的辅助。如果研究对象是动物或者植物,而没有可以利用的组学数据,那怎么办?
无注释数据时,可以选择 CAVIAR、CAVIARBF、SNPtest等方法。不过,这些方法用了穷举搜索,速度上堪忧。
这里推荐 FINEMAP,它可以用于:
- 识别因果SNP(causal SNP)
- 估计因果SNP的效应大小
- 估计因果SNP的遗传贡献
FINEMAP 的统计模型类似于 CAVIAR 和 CAVIARBF,但算法上有一个很大的区别。FINEMAP 使用shotgun stochastic search(SSS)算法,与 CAVIARBF 的穷举搜索算法相比速度提高了非常多,避免计算量大的穷举搜索。PAINTOR 也可在无注释数据时使用,但相比而言 FINEMAP 的结果会更准确。
另外,FINEMAP 有一个方便的地方是,在设定好最大causal SNPs 数量 k 后,得到的结果包含了 1 到 k 个不同 causal SNPs 的后验概率以及各个 causal SNPs 数量的概率,非常方便进行后续的分析。
下载
可在 http://www.christianbenner.com 根据自己的系统平台选择编译好的文件。以 Linux 为例:
wget http://www.christianbenner.com/finemap_v1.4_x86_64.tgz
tar -xzvf finemap_v1.4_x86_64.tgz
解压后文件夹中的 finemap_v1.4_x86_64 可直接执行。
输入文件
FINEMAP 的输入需要 master 文件,z 文件和 ld 文件。如果有 5 个不同的显著位点,那么需要 1 个 master 文件和 5 个不同的 z文件 和 ld 文件。
另外,如果有每个 SNP 的先验概率,则可以提供对应的 k 文件。k 文件属于可选项,不提供的话 FINEMAP 会假定每个 SNP 的先验概率为 1。如果想要更准确,并且研究对象是人类,推荐使用 PolyFun 利用功能注释数据计算出先验概率,提供给 FINEMAP 让 fine-mapping 结果更可靠。这篇教程介绍的是无功能注释数据时的 fine-mapping,不会对 PolyFun 的使用进行介绍。
因为每个人的数据格式不太一样,这里仅说明每个文件的格式要求,操作的时候需要根据实际情况自己写脚本生成相应文件。
Z file
一个用空格分隔的文本文件,包含需要分析区域的 GWAS summary statistics 信息,每行一个SNP。文件必须包含列名,每一列分别是:
rsid:SNP 名字
chromosome:染色体名称,性染色体的名字与 precomputed SNP correlations 文件保持一致即可
position:物理位置
allele1:effect allele,在 FINEMAP 中特指 minor allele
allele2:reference allele,在 FINEMAP 中特指 major allele
maf:minor allele (allele1)频率
beta:GWAS结果的 effect sizes
se:the standard errors of effect sizes
例子:
rsid chromosome position allele1 allele2 maf beta se
rs1 10 1 T C 0.35 0.0050 0.0208
rs2 10 2 A G 0.04 0.0368 0.0761
rs3 10 3 G A 0.18 0.0228 0.0199
LD file
一个用空格分隔的文本文件,当中是SNPs的相关性矩阵(correlation matrix)。FINEMAP 官方文档建议计算皮尔逊相关系数,不过文献中未特别指出矩阵的计算方法。如果要方便点,也可以用 plink 的 --r square 计算出 r matrix,效果差不多。
例子:
1.00 0.95 0.98
0.95 1.00 0.96
0.97 0.96 1.00
注意,SNP 顺序必须与 Z 文件中的顺序保持一致
Master file
分号分隔的文本文件,不包含空格。这个文件记录的是数据集和配置参考。第一行是列名,后续每一行是一个数据集和对应的参数。文件每一列分别是:
z:Z 文件的名称(输入)
ld:LD 文件的名称(输入)
bcor:BCOR 文件的名称(输入)
snp:结果输出 SNP 文件的名称(输出)
config:结果输出 CONFIG 文件的名称(输出)
cred:结果输出 CRED 文件的名称(输出)
n_samples:GWAS 样本数量
k:K 文件的名称(可选输入,可忽略)
log:LOG文件的名称(可选输出,可忽略)
ld 列和 bcor 列二选一,如果 ld 信息是用纯文本表示的矩阵,填入 ld 文件即可。
举例,分别计算两个数据集的 master 文件如下所示:
z;ld;snp;config;cred;log;n_samples
dataset1.z;dataset1.ld;dataset1.snp;dataset1.config;dataset1.cred;dataset1.log;5363
dataset2.z;dataset2.ld;dataset2.snp;dataset2.config;dataset2.cred;dataset2.log;5363
运行
以程序中的示例数据为例:
运行 shotgun stochastic search 方法的 fine-mapping:
./finemap_v1.4_x86_64 --sss --in-files example/data --dataset 1
–infiles 是 master 文件。 –dataset 接的是 master 文件中的行号,行号从 1 开始,填入 1 代表使用第 1 行的 z 文件、ld 文件和配置参数进行分析。
除了 shotgun stochastic search,还可以使用 stepwise conditional search 方法,它的逐步条件处理过程类似于 GCTA COJO 中的实现:
./finemap_v1.4_x86_64 --cond --in-files example/data --dataset 2
如果想要了解指定 SNP 是否 causal,可以:
./finemap_v1.4_x86_64 --config --in-files example/data --dataset 1 --rsids rs30,rs11
结果解读
输出结果有 .snp、.cred、.config 三种不同后缀的文件。
.config 记录了分析时选用的参数。
.snp 文件是 model-averaged posterior summaries,每一行是一个 SNP。在假定不同 causal SNPs 数量时会得到不一样的结果,而这个文件包含了所有结果的摘要情况。
.cred 文件最后会带有一个数字,这个数字代表的是 causal SNPs 数量 k。比如,.cred5 是设定 causal SNPs 为5个而得到的计算结果。这个文件中,包含了数量 k 的后验概率和和推断出的 causal SNP 后验概率。
如果对 FINEMAP 的方法不是特别了解,可以只看 .cred 文件中的结果,从中选出 k 值和相应的 causal SNPs。
总结
FINAMAP 的速度非常快。经过实践,在 6000 个样本的情况下,截取 top SNPs 上下游各 3Mb 区域进行分析,设定最大 causal SNPs 数量为 10,每一个区域的计算时间大概 20 分钟,内存占用非常小。在显著位点很多的时候,FINEMAP 的优势体现出来了。
![图片[2]-FINEMAP:使用GWAS摘要数据进行无功能注释数据的精细定位(Fine-mapping) - 生物信息-实验盒](https://www.shiyanhe.com/wp-content/uploads/2020/07/wx-banner-smallsize.jpg)